



meerkat
-
Posts
236 -
Joined
-
Last visited
-
Days Won
9
Content Type
Profiles
Forums
Events
Gallery
Downloads
Blogs
Media Demo
Store
Everything posted by meerkat
-
I accept that the time taken for water addition can affect the taste and appearance of the product, but I doubt that this is because of any chemical reactions taking place with the water. I know through personal experience, as well as from studying the theory, that some of the cogeners are not soluble in water. If you add the water too quickly there will be zones in the mixing vessel where the cogeners are in contact with high concentrations of water and very little alcohol. The oils can form emulsions that cause haze and taste concentrations, and these emulsions are extremely difficult to get back into solution. In his book "Distillery Operations" Payton Fireman refers to a blending operation where he weighed out the required quantity of water and then added the whiskey to it. The batch was ruined and had to be entirely re-distilled. The first whiskey to enter the water would have quickly become highly diluted, exactly as I have described above, and the oils would have come out of solution. So I don't believe the problem is a chemical reaction that is happening over time. It seems to me to be a physical phenomenon where the oils are "squeezed" out of the alcohol and impact the appearance and taste. This physical phenomenon would be dependent not so much on the time that it takes to add the water, but rather the local rate at which it was added. Ideally the water should be added via a sparger where there will be many zones of low water concentration during mixing, rather than only one zone of much higher water concentration. The blend should be stirred during the entire operation. If my understanding is correct, the reason for better tasting products being produced when the dilution is done over a long period would be because the rate at which the water was added was much lower than when it was done quickly. It would be interesting to hear from @JustAndy whether the water was added at a lower rate when it was done over weeks rather than over one or two days.
-
It is important to remember that the temperature is not an independent variable that we can set arbitrarily. The temperatures in the table I posted earlier are boiling point temperatures and are fixed by the composition of the boiling liquid. The only way to change the boiling point for a given concentration is to change the pressure, but I am assuming here that everything is being done at normal atmospheric pressure. Let me take the data from the 4th row in the table as an example and assume you have material from a previous stripping run at 32.26 %ABV. If you put this in a pot still and start heating it, the temperature will rise but it will not boil until the temperature reaches 85.3°C. Spirit at this composition can only be distilled at 85.3°C and at any lower temperature there will be no boiling and therefore no distillation. If you continue heating the pot and boiling the spirit the boiling point will slowly rise as the concentration of the ethanol in the pot decreases - because more ethanol than water has been removed by distillation. The new temperature is just an indicator of the new composition in the pot, and cannot be increased (or decreased) arbitrarily while maintaining boiling. Even when we use reflux on a column to "control the temperature" we are not truly controlling the temperature as an independent variable. Changing the reflux rate changes the composition in the column and the measured temperature is just an indication of that changed composition. The measured temperature can be used to interpolate the data in my table to work out what the actual composition is because we know it is at its boiling point. We are all guilty of talking of controlling the temperature, but strictly that is not true. The temperature is just a proxy for the composition. We could take this analogy a step further and say we do not really even measure the temperature. Just as we have no direct way to measure the composition inside the column, we actually have no way to directly measure its temperature. We measure the length of a column of mercury in a pencil thermometer (or the resistance of an RTD probe) and from this length (or resistance) we infer a temperature. And in turn from this inferred temperature we infer a composition. I have heard of distillers preferring "slow distillation" but have no direct experience of this myself. True distillation is not impacted by the rate at which it occurs (as long as the column is still operating properly) so if the taste/smell of the product changes with the rate of distillation there is some other phenomenon occurring. We know that in alcohol distillation there are some chemical reactions going on - particularly between any sulfur compounds and the copper - so I can accept that the rate at which a product is distilled can affect its quality but it is not the distillation itself that is having that effect.
-
In general, mixtures of two liquids will boil at temperatures between the boiling points of the two pure liquids and the boiling point will vary with the concentration. The ethanol-water mixture is a bit different in that it forms an azeotrope. From the first sentence above we would expect the boiling point (at atmospheric pressure) of a mixture of ethanol and water to be between 100°C (boiling point of pure water) and 78.37°C (boiling point of pure ethanol). In most cases this is true. However, a mixture containing 95.58 mass % ethanol will boil at 78.15°C, which is lower than the boiling point of pure ethanol. This is called the azeotrope. To really split hairs, it is called a minimum boiling azeotrope because you can also get maximum boiling azeotropes where the boiling point of the mixture is higher than either of the pure boiling points. The existence of the azeotrope is why we cannot achieve 100% ethanol by normal distillation. The lowest temperature occurs at the top of the column and for ethanol-water this would be the azeotrope temperature of 78.15°C and no matter how much taller you made the column you could never go beyond the 95.58 mass % concentration. I have attached a table of boiling point data. In addition to showing the boiling point at various liquid concentrations it also shows the composition of the vapour that is generated. Between 100°C and the azeotrope ethanol is more volatile than water and there will be a higher concentration of ethanol in the vapour than was in the boiling liquid. If this were not so, distillation columns would not work. The "VLE" in the title on the attachment stands for Vapour Liquid Equilibrium - sorry for the jargon. Carey and Lewis MF Mass and ABV.pdf
-

Anyone running continous stills? Really suprised at the lack of them.
meerkat replied to Mverick160's topic in Technique
For a column of 300 mm ID you definitely do not want to go for the split flow design you have shown. It is unnecessarily complex and restricts the bubbling area. I have seen single pass trays of 2 m diameter working very well, even back in the days of bubble caps. I have never seen a downcomer with perforations at the bottom. You want the flow down the downcomer to be as unimpeded as possible, especially if the liquid is not totally clear. I would leave the bottom of the DC totally open. The residence time is calculated as DC volume divided by volumetric flowrate. Your flowrate of 480 l/h (actually a bit less because some goes out as vapor) is equivalent to 0.000133 m3/s going down the DC and for an 80 mm ID pipe 300 mm long the volume of the DC is 0.0015 m3. If you divide m3 by m3/s the m3 cancels and you are left with seconds, so that is why it is called a residence time. Here we get 11.3 seconds - a nice safe number. The downcomer does not run full. Typically the level in the downcomer would be 30 to 50 % of the tray spacing. So the true time that the liquid spends in the downcomer is 3 to 5 seconds, but this is enough for the bubbles to disengage, thus avoiding vapor being carried downwards when it should be going upwards. The reason the level in the downcomer backs up is because the pressure on any tray has to be a bit higher than on the tray above it to force the vapor up through the tray and this pressure holds the liquid in the downcomer back. There is also a small pressure drop as the liquid flows under the downcomer and onto the tray. The diagram below, from Peters and Timmerhaus, shows a variety of different tray types but I like these simple downcomers Here they have shown segmental downcomers where the column shell forms the outer part of the downcomer but this is difficult to fabricate in smaller columns and it is more usual to weld a pipe or D section into the tray to achieve the all-round seal. A very important aspect shown in this diagram is the sealing of the bottom downcomer in the base of the column. For a tray to function properly the vapor must not flow up any of the downcomers. The bottom tray seals first and then the seal is achieved in turn on each higher tray until all the DCs are sealed. Imagine the column at start-up with the bottom DC sealed by the liquid in the pot. When boiling starts in the pot the vapor cannot flow up the bottom DC and it flows up through the perforations in the first tray. The bottom of the DC from the 2nd tray will not be sealed with liquid yet and vapor will flow up this DC, as well as through the perforations of the second tray. But because the vapor flowing up through the bottom tray prevents any liquid from weeping through the holes the liquid will accumulate on the bottom tray until it can overflow the weir into the downcomer. As long as the downcomer projects above the tray more than the gap between the tray and the bottom of the DC from tray 2, when the liquid gets to a height sufficient to overflow into the downcomer it will have sealed the bottom of the next downcomer. Now all the vapor from tray 1 goes through the perforations in tray 2 and the same process allows the next downcomer to achieve its seal. 30 mm is a reasonable height for the weirs, but maybe a bit on the high side. They cannot be too low because (as explained above) they must be higher than the gap at the bottom of the downcomer so that the tray can seal the bottom of the DC. The higher the weir, the higher the liquid level will be on the tray. The higher this level the higher the efficiency of the tray, but also the more easily the tray will weep. It's all a trade-off between the competing factors.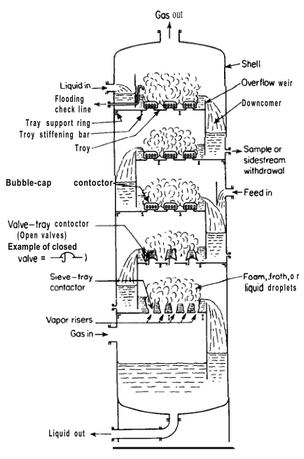
-
Anyone running continous stills? Really suprised at the lack of them.
meerkat replied to Mverick160's topic in Technique
@Modernity I would be wary of using perforated trays if you are going to be distilling grain in. If you are going to make the trays into a removable cassette then that could work if you need to clean them. Maybe wait for @PeteB to post his photos and diagrams to see how he did it. He and I discussed using very simple splash trays before he built his first column, and he came up with a very innovative way of installing the trays but I have not seen what he finally built. If you do go with the perforated trays be generous with your downcomer sizing. I like to allow a downcomer residence time of 10 seconds, based on the full volume of the downcomer. If your tray spacing is around 300 mm then it would be better to install a 3” downcomer. I also prefer D shaped downcomers. You can make these easily by splitting a 6” pipe in half longitudinally and then welding in a plate to seal the straight part of the D. Half a 6” pipe has double the area of a 3” pipe, but is still only 3” wide. Some references for downcomer design also specify a maximum velocity, but this only comes into play with large hydrocarbon columns that can have +3 ft tray spacings. The hole area (perforations) is much harder to calculate. I found this reference that states % Hole Area: This is the ratio of hole area to bubbling area. The default practice is to target a hole area of 8 to 10 % of bubbling area for pressure services. The acceptable range for percentage hole area is 5 % to 15 %. However for some critical services, we can go % hole area up to 17-17.5 % provided that weeping is under control. Hole areas below 5 % are not used. Despite their claim that hole areas of less than 5 % are not used I designed a column that uses 2.9 % and has been running very stably 24/7 for 33 years. That column was a bit unusual in that it had a very high liquid to vapor ratio and I had a lot of pressure drop to play with. For your stripper you will also have a relatively high liquid to gas ratio and I would guess that it will need 6 to 8 % open area, but that really is just a guess. If the hole area is too small it will unnecessarily limit the capacity of the column, and if the hole area is too large the trays will weep and their separation power will be low. Rather than taking the risk of deciding on the hole area yourself it might be better to buy trays from a supplier with a track record and who offers a guarantee. (I am not touting for business – I do not supply equipment or consulting services.) -
Hand Sanitizer Hydrometer Reading offset?
meerkat replied to Jedd Haas's topic in Hand Sanitizer Production
@John Bassett Yes, that is correct. The final product should have an apparent proof of 157.8 if its true proof is 160. -
Hand Sanitizer Hydrometer Reading offset?
meerkat replied to Jedd Haas's topic in Hand Sanitizer Production
This post is just to pull this thread to the top again. I have made an edit to the obscuration factor that I had calculated above for glycerol and I would like anyone who has used the old number to be aware of the change. @John Bassett @CalwiseSpirits @Jedd Haas @DrDistillation @SCLabGuy -
Hand Sanitizer Hydrometer Reading offset?
meerkat replied to Jedd Haas's topic in Hand Sanitizer Production
@SCLabGuy - I am happy to share the calculations for the obscuration numbers, but I doubt whether many would be interested to see them here. They are hand-written and I will send you a scan if you write to me at the support address given in my software. The calculations in AlcoDens LQ are based on hard data and not on theoretical calculations. This is important for the contraction calculations, which are totally neglected in the simple obscuration estimates I have done for the glycerol. I would be reluctant to add untested calculations into the software. And I get too many people saying that the software needs to be simplified for me to go the other way and add in extra complications and options. Thanks for the pointer to the FDA guidelines. -
Hand Sanitizer Hydrometer Reading offset?
meerkat replied to Jedd Haas's topic in Hand Sanitizer Production
Hi @Jedd Haas - please can you point me to the FDA recommendation that the alcohol content should be verified with a hydrometer. I have not been following the sanitizer threads closely and am not up to speed on the regulations. Using a hydrometer to check the final product will indicate if there are gross mistakes, but it is not sufficient to truly verify the composition of the product. It will tell you if your formulation is wrong, but will not confirm if it is correct. Here are a few of my observations that may help you good people helping us all out by providing the much-needed sanitizers. Isopropanol has a very similar density to ethanol. My estimate is that if you substitute 5 parts by volume of the ethanol in a 160 proof spirit with 5 parts of isopropanol then the apparent proof would increase to 160.16. In a non-potable product I would guess this is negligible. The glycerol and peroxide are both heavier than ethanol or water, so they will definitely obscure the ethanol. Unfortunately both are lighter than sugar, so I can't push my blending calculator here! I have tried to work out obscuration rules for glycerol and peroxide in the same format that the TTB uses for sugar. The TTB uses the units of 100 mg of sugar per 100 ml of spirit. This is equivalent to 1000 mg of sugar per 1000 ml of spirit or 1 gram per liter, which I find a much easier unit to work with, so I will quote everything in those terms, but the numbers are actually the same as what the TTB uses. [Edit April 10: After doing more calculations together with @SCLabGuy we found that the numbers below were a bit too high. My latest number for glycerol (the only bit that really counts) is 0.12 Proof per gram/L. This makes it even more reasonable to use a hydrometer to check the final product - as the FDA recommends. End Edit] For glycerol I come to 0.23 Proof per gram/L and for peroxide 0.29 Proof per gram/L. These would be specifically for an 80 % ABV alcohol content and would definitely vary with the alcohol strength. These numbers compare with the 0.4 Proof per gram/L the TTB gives for sugar in 80-100 proof spirits. Glycerol at 1.45 v/v is equivalent to 18.3 gram/L so the obscuration would be 18.3 x 0.23 = 4.2 Proof. Peroxide at 0.125 v/v is equivalent to 0.181 gram/L, making the obscuration 0.181 x 0.29 = 0.05 proof and probably not worth worrying about. So if we ignore the variance caused by the isopropanol and the peroxide we can say that the apparent proof should be 160 - 4.2 = 155.8 Proof. Some formulations recommend half the glycerol (i.e. 0.725 v/v) and then the apparent proof would be 160 - 2.1 = 157.9 Proof. Please note that these are all theoretical calculations and you should verify everything with actual measurements. I look forward to @SCLabGuy showing his lab results to see how close we can get with calculations. If my numbers need to be expressed in different terms to make them more useful in practice please let me know and I can rework them. -
I haven't seen a way to run Windows applications on the iPad itself, but there are several solutions which allow you to use the iPad to view and control a Windows program running on a separate Windows computer. Have a look at Team Viewer, Jump Desktop, Parallels Access etc. But please note, we have not tested any of these ourselves.
-
An AlcoDens user recently informed me that he found an even better way to run AlcoDens on a Mac. He is using CrossOver from CodeWeavers. This software costs about US$ 50.00 but it makes the installation of AlcoDens so much easier. It isn't free like the Wine option, but it's a whole lot cheaper and more convenient than having to run a separate Windows machine just for AlcoDens. I have updated the AlcoDens for Mac page with the links to CrossOver and some more details on how to do the installation. CrossOver is also available for Chromebook and for Linux, but I haven't tested those.
-
Each plate is at a slightly *lower* temperature than the tray below it. In a typical vodka column the base would be at >100°C (212°F) and the top at around 80°C (176°F) - but these will vary with time in a batch column. The way to understand the temperatures is that boiling causes the more volatile component (ethanol) to move to the top of the column while the less volatile component (water) concentrates at the base of the column. The top product containing more ethanol than the base product will have a lower boiling point than the base product. And each plate will contain a slightly higher concentration of ethanol than the tray below it and therefore be at a slightly lower temperature than that tray. When we control the heat input or reflux on a column it seems as though we are controlling the temperature, but in fact we are controlling the composition and the change in temperature is a *result* of the change in composition. It may seem that the change in temperature is causing the change in composition but it is really the other way around. You cannot control the temperature of each tray individually. As you change the heat input or reflux the entire temperature profile in the column will change. We use temperature as the parameter to measure because it is the fastest and cheapest measurement to make to indicate the composition, which is what we really want to control. Another important factor in large vodka columns is that any change in composition profile across all the plates will result in different temperature changes on different plates. This means that the temperature swing will be much higher on some plates than on others, so we can use these sensitive plates to measure the temperature and get the quickest indication of a change in composition profile. So you get the seemingly strange situation where the reflux to the top plate is controlled by the temperature 2/3 of the way down the column. The reason for washing with water is complex. Basically, the *relative* volatility of different alcohols changes according to the amount of water present. By increasing the amount of water you can cause a higher alcohol (eg amyl alcohol or fusel oil) which would normally be less volatile than ethanol and therefore concentrate near the base of the column to increase in relative volatility and therefore migrate to the top of the column in higher quantities. The calculations are horrendous and even the most powerful computer simulations struggle with the hydroselection column. When I have had to design these columns I simply took the number of trays and water feed ratio from plants that worked well, and limited my design to getting the plate hydraulics optimized for capacity and pressure drop.
-
Jim, you have opened a can of worms. But they are all very small worms, so mostly they can be ignored. Strictly speaking, SG is not the same thing as grams per ml. For most purposes they can be taken to be the same, but SG is actually the mass of 1 ml of the fluid divided by the mass of 1 ml of water - both at 60°F. The mass of 1 ml water at 60°F is 0.9990 gram so 1 ml of 80 Pf has a mass of 0.9518 x 0.9990 = 0.95085 g at 60°F. This makes its density 0.95085 g/ml. The value of 0.12616 gallons per pound in Table 4 is measured in air. This tells you what volume is occupied by alcohol that weighs 1 lb. But normal density tables (not TTB tables) are based on mass, not weight, so we have to convert the 0.12616 gall per lb weight to 0.12602 gallon per lb mass. Your conversion procedure was correct, so we can convert this directly to a density using your formula : 1/ (0.12602 * 3785.41 ml/gal / 453.592 g/lb)=0.95085. Same as above. Do your scales give "in air" values, or are they corrected to absolute mass? As you pointed out, does it really matter?
-
You can have plates (or packing) in continuous stills and in batch stills. The purpose of the plate (or packing) is to increase the enrichment or rectification. Basically these plates emulate the repeated pot still distillations you suggested in your first post. I suspect this is why Southernhighlander said he can do 21 distillations in one run - 1 for the pot plus 20 plates. In a column the stages are simply stacked on top of each other rather than being separated in time (as they would be with repeated pot still runs). The difference between a continuous still and a batch still is that (theoretically) all the strengths remain constant in a continuous still, but will change with time in a batch still. In a large continuous plant the columns will run 24/7 - always receiving the same strength feed and delivering the same strength product. I have never seen a single *continuous* column capable of producing neutral spirit. Typically there would be at least 4 continuous columns in series to make neutral spirit. The first column is the stripper and it produces about 30-40 ABV spirit. The second column is a rectifier that will make "industrial grade" 95 ABV alcohol. Heads are removed from each of these first 2 columns. The spirit from the 2nd column is diluted down to around 15 ABV with clean water and fed to the hydroselection column. This is an "upside-down" column and the product comes off the bottom and is taken to the final rectifier where it is enriched to 95-96 ABV. Beyond these 4 columns there could be 2 or 3 more columns for low grade recovery, de-methylation etc. You can (thousands of distillers do) make neutral spirit in a single *batch* still of the type described by Southernhighlander. The difference between doing it in a single column batch-wise and doing it continuously in multiple columns is that you will get a higher percentage of your alcohol out as neutral grade in a continuous setup compared with selecting only the best "hearts" from the batch still. It is of course far easier to run continuous stills using automatic instruments than it is to run a batch column.
-
If you think the world is over-reacting to COVID-19 you have not understood the problem. Comparing it to normal flu is irrelevant. You need to compare it to the 1918 Spanish Flu. The only good news so far is that the Chinese have proven that it can be beaten - using basically the same techniques that worked in 1918 and against SARS. If you are prepared to invest half an hour into understanding the process and the risks we face, have a read through this article. Our problem in the west is that we rank personal freedoms above those of the group and this makes it more difficult for us to implement the group focused solutions that have worked for the Chinese.
- 2 replies
-
- 2
-

-
- insurance
- insuranceman 2.0
- (and 4 more)
-
Hi Craill, Welcome to the forum from a fellow South African. I'm sorry I can't help you with any CAD drawings, but I'm sure someone here will. Regards meerkat
-
All blending for flavor is by trial and error, but this excellent suggestion means that you are only trialing one variable (flavor) while the other variable (proof) is already solved. Simple, but ingenious!
-
A safety factor of 5% is excessive. Using the TTB tables or software like AlcoDens you can do these calculations exactly without creeping up on the final answer with these large safety factors. Even if you totally disregard the shrinkage on mixing in your calculations when proofing down from 93 to 25 abv your volume would be out by only 1.8 %, but of course your proof would be wrong too. If it is the temperature change that you are worried about, you need a temperature swing of around 100 F to get a 5% change. There is a free trial version of AlcoDens available. Please give it a try and I am sure you will save yourself a lot of time and frustration.
-
It's probably not fully on-topic, but the only relevant feedback that I have had from users of my blending software is that some time is required between the physical blending process and when it is possible to get an accurate and stable proof reading. The most plausible reason I have heard for this is that micro-bubbles of air are entrained into the spirit during mixing and affect the spirit density, and these bubbles take some time to escape. The typical duration given for this "settling" period is 12 to 24 hours. Nobody has ever suggested to me that 3-4 days are required and I suspect that this bubble-removal process is not what you are asking about.
-
I found some data on the web that gives the particle density of corn as 1.50 g/cm3. This converts to a displacement of 0.080 gallon per pound. Your example of 250 lbs would give a displacement of 250 x 0.080 = 20 gallons, so theoretically you would need to add 80 gallons of water to get to 100 gallons total. The data mentioned above points out that densities vary with moisture content etc, so you should take the value of 0.080 gall/lb as indicative only and experimental values will be best.
- 1 reply
-
- 3
-

-
@Still_HollerSorry to drag up such an old thread, but I was grappling with this recently and came upon this thread while searching for the answer. I believe I have found the answer in 27 CFR 30.71.. See also 30.72. § 30.71 Optional method for determination of proof for spirits containing solids of 400 milligrams or less per 100 milliliters. The proof of spirits shall be determined to the nearest tenth degree which shall be the proof used in determining the proof gallons and all fractional parts thereof to the nearest tenth proof gallon. The proof of spirits containing solids of 400 milligrams or less per 100 milliliters shall be determined by the use of a hydrometer and a thermometer in accordance with the provisions of § 30.23. However, notwithstanding the provisions of § 30.31, the proprietor may, at his option, add to the proof so determined the obscuration determined as prescribed in § 30.32.
-
@ZimDist That depends on how clean you want to make it, and how much you can sell it for. In a liquor store you will probably find a 4 to 1 ratio between the most expensive and the cheapest vodkas. What is viable at the top end is not viable at the bottom end. I've read of expensive vodkas being filtered through beds of diamonds. Maybe that is viable in Zimbabwe? You certainly could put it in a batch still and take off some more heads at 95+ abv (ideally 96+). There are threads here that discuss the equipment required to achieve 190+ proof. It would require a bit of experimentation to get the optimum balance between the cost of losses (as additional heads taken off) and the improvement in quality. The neutral cane spirit that you can buy ex SA is made in continuous plants where there are multiple columns with multiple side take-offs in addition to the heads. I cannot see continuous distillation being financially viable at below 10,000 liters per day (probably more like 40 kl/day).
-
The same convention is used in South Africa. There is very little grain-derived potable alcohol available here. It is all grape or sugar cane based. What we call cane spirit here is really a neutral rum. Locally vodka is made from the same cane-based neutral spirit. Each bottler has their own "magic" that converts cane to vodka, most of it involving treatment with activated carbon. I don't know how much of it is hype and how much is valid technology. Some very well-known international vodka brands are made in South Africa from cane spirit. If you want to make a very smooth vodka, you must start with a very smooth cane spirit. In my experience it is difficult to remove harshness from a spirit by any method other than distillation.
-
@joshYPV Yes, it is ideal for that. As long as you know (or can measure) the sugar content of your honey you can treat it as syrup in the software. If you need some help in setting up the calculation send me an email using the support address listed in the software, with a typical proofing calculation and I will set it up for you.
-
I was looking at the Hillbilly Stills calculator linked by @Thatch to try to understand why their numbers were so different from mine. The main difference between the calculations is that I had 2.5 %abv left in the still heel, but the Hillbilly calculation would give 0.5% in the heel. My calculation assumed that you were using a pot still without any trays or reflux, and then worked back from the 20 %abv ending strength in the parrot to get the heel strength. But then I calculated what strength you would get in the parrot at the start of the run and it would be impossible to get 82 %abv from a 8.5 %abv mash. I guess this means that you are using some trays and reflux in your still. This is one of the difficulties in designing a calculator - every still is slightly different. I can see that the Hillbilly calculator would be very useful, so I tried to understand what it is actually doing. It seems that it is based on the assumption that 95% of the alcohol in the original mash is recovered in the distillate. Because of the virtual impossibility of being able to model every possible still configuration, to be able to calculate the volume split between the distillate and the heel you need 4 bits of information. These are the volume and strength of the initial mash, and the strengths of the distillate and of the heel. The Hillbilly calculation asks for only the first 3 items and it calculates the heel strength internally by assuming that 95 % of the alcohol is recovered as distillate. If it is reasonable to be able to estimate the 4 items I have listed, it would be very easy to make a spreadsheet to calculate the quantity of distillate and remaining heel.



